Metagenome Binning
Overview
Teaching: 50 min
Exercises: 10 minQuestions
How can we obtain the original genomes from a metagenome?
Objectives
Obtain Metagenome-Assembled Genomes from the metagenomic assembly.
Check the quality of the Metagenome-Assembled genomes.
Metagenomic binning
Original genomes in the sample can be separated with a process called binning. This process allows separate analysis of each species contained in the metagenome with enough reads to reconstruct a genome. Genomes reconstructed from metagenomic assemblies are called MAGs (Metagenome-Assembled Genomes). In this process, the assembled contigs from the metagenome will be assigned to different bins (FASTA files that contain certain contigs). Ideally, each bin corresponds to only one original genome (a MAG).
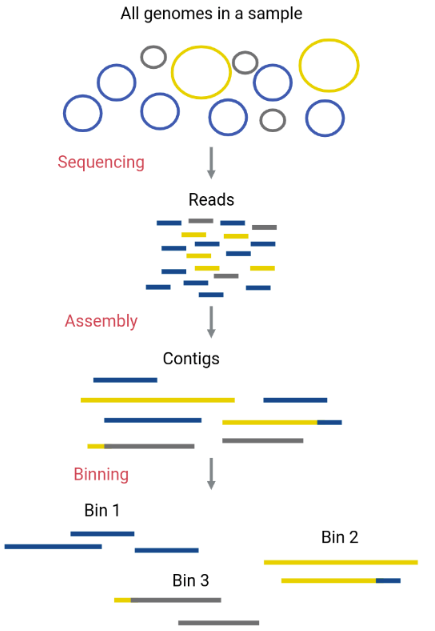
Although an obvious way to separate contigs that correspond to a different species is by their taxonomic assignation, there are more reliable methods that do the binning using characteristics of the contigs, such as their GC content, the use of tetranucleotides (composition), or their coverage (abundance).
Maxbin is a binning algorithm that distinguishes between contigs that belong to different bins according to their coverage levels and the tetranucleotide frequencies they have.
Discussion 1: Relation between MAGs and depth
The sequencing center has returned you a file with 18,412 reads. Given that the bacterial genome size range between 4Mbp and 13Mbp (Mb=10^6 bp) and that the size of the reads in this run is 150bp. With these data, how many complete bacterial genomes can you reconstruct?
Solution
None, because 18,412 reads of 150bp give a total count of 2,761,800bp (~2Mbp). Even if no read maps to the same region, the amount of base pairs is inferior to the size of a bacterial genome.
Let us bin the sample we just assembled. The command for running MaxBin is run_MaxBin.pl, and the arguments it needs are the FASTA file of the assembly, the FASTQ with the forward and reverse reads, the output directory, and the name.
$ cd ~/dc_workshop/results/assembly_JC1A
$ mkdir MAXBIN
$ run_MaxBin.pl -thread 8 -contig JC1A_contigs.fasta -reads ../../data/trimmed_fastq/JC1A_R1.trim.fastq.gz -reads2 ../../data/trimmed_fastq/JC1A_R2.trim.fastq.gz -out MAXBIN/JC1A
MaxBin 2.2.7
Thread: 12
Input contig: JC1A_contigs.fasta
Located reads file [../../data/trimmed_fastq/JC1A_R1.trim.fastq.gz]
Located reads file [../../data/trimmed_fastq/JC1A_R2.trim.fastq.gz]
out header: MAXBIN/JC1A
Running Bowtie2 on reads file [../../data/trimmed_fastq/JC1A_R1.trim.fastq.gz]...this may take a while...
Reading SAM file to estimate abundance values...
Running Bowtie2 on reads file [../../data/trimmed_fastq/JC1A_R2.trim.fastq.gz]...this may take a while...
Reading SAM file to estimate abundance values...
Searching against 107 marker genes to find starting seed contigs for [JC1A_contigs.fasta]...
Running FragGeneScan....
Running HMMER hmmsearch....
Try harder to dig out marker genes from contigs.
Marker gene search reveals that the dataset cannot be binned (the medium of marker gene number <= 1). Program stop.
It seems impossible to bin our assembly because the number of marker genes is less than 1. We could have expected this as we know it is a small sample.
We will perform the binning process with the other sample from the same study that is larger. We have the assembly precomputed in the ~/dc-workshop/mags/ directory.
$ cd ~/dc_workshop/mags/
$ mkdir MAXBIN
$ run_MaxBin.pl -thread 8 -contig JP4D_contigs.fasta -reads ../data/trimmed_fastq/JP4D_R1.trim.fastq.gz -reads2 ../data/trimmed_fastq/JP4D_R2.trim.fastq.gz -out MAXBIN/JP4D
It will take a few minutes to run. Moreover, it will finish with an output like this:
========== Job finished ==========
Yielded 4 bins for contig (scaffold) file JP4D_contigs.fasta
Here are the output files for this run.
Please refer to the README file for further details.
Summary file: MAXBIN/JP4D.summary
Genome abundance info file: MAXBIN/JP4D.abundance
Marker counts: MAXBIN/JP4D.marker
Marker genes for each bin: MAXBIN/JP4D.marker_of_each_gene.tar.gz
Bin files: MAXBIN/JP4D.001.fasta - MAXBIN/JP4D.004.fasta
Unbinned sequences: MAXBIN/JP4D.noclass
Store abundance information of reads file [../data/trimmed_fastq/JP4D_R1.trim.fastq.gz] in [MAXBIN/JP4D.abund1].
Store abundance information of reads file [../data/trimmed_fastq/JP4D_R2.trim.fastq.gz] in [MAXBIN/JP4D.abund2].
========== Elapsed Time ==========
0 hours 6 minutes and 56 seconds.
With the .summary file, we can quickly look at the bins that MaxBin produced.
$ cat MAXBIN/JP4D.summary
Bin name Completeness Genome size GC content
JP4D.001.fasta 57.9% 3141556 55.5
JP4D.002.fasta 87.9% 6186438 67.3
JP4D.003.fasta 51.4% 3289972 48.1
JP4D.004.fasta 77.6% 5692657 38.9
Discussion: The quality of MAGs
Can we trust the quality of our bins only with the given information? What else do we want to know about our MAGs to use for further analysis confidently?
Solution
completeness is fundamental to know which data you are working with. If the MAG is incomplete, you can hypothesize that if you did not find something, it is because you do not have a complete genome. Genome size and GC content are like genomic fingerprints of taxa, so you can know if you have the taxa you are looking for. Since we are working with the mixed genomes of a community when we try to separate them with binning, we want to know if we were able to separate them correctly. So we need to measure contamination to know if we have only one genome in our bin.
Quality check
The quality of a MAG is highly dependent on the size of the genome of the species, its abundance in the community and the depth at which we sequenced it. Two important things that can be measured to know its quality are completeness (is the MAG a complete genome?) and if it is contaminated (does the MAG contain only one genome?).
CheckM is an excellent program to see the quality of our MAGs. It measures completeness and contamination by counting marker genes in the MAGs. The lineage workflow that is a part of CheckM places your bins in a reference tree to know to which lineage it corresponds and to use the appropriate marker genes to estimate the quality parameters. Unfortunately, the lineage workflow uses much memory, so it cannot run on our machines, but we can tell CheckM to use marker genes from Bacteria only to spend less memory. This is a less accurate approach, but it can also be advantageous if you want all of your bins analyzed with the same markers.
We will run the taxonomy workflow specifying the use of markers at the domain level, specific for the rank Bacteria,
we will specify that our bins are in FASTA format, that they are located in the MAXBIN directory
and that we want our output in the CHECKM/ directory.
$ mkdir CHECKM
$ checkm taxonomy_wf domain Bacteria -x fasta MAXBIN/ CHECKM/
The run will end with our results printed in the console.
--------------------------------------------------------------------------------------------------------------------------------------------------------
Bin Id Marker lineage # genomes # markers # marker sets 0 1 2 3 4 5+ Completeness Contamination Strain heterogeneity
--------------------------------------------------------------------------------------------------------------------------------------------------------
JP4D.002 Bacteria 5449 104 58 3 34 40 21 5 1 94.83 76.99 11.19
JP4D.004 Bacteria 5449 104 58 12 40 46 6 0 0 87.30 51.64 3.12
JP4D.001 Bacteria 5449 104 58 24 65 11 3 1 0 70.48 13.09 0.00
JP4D.003 Bacteria 5449 104 58 44 49 11 0 0 0 64.44 10.27 0.00
--------------------------------------------------------------------------------------------------------------------------------------------------------
To have these values in an output that is more usable and shearable, we can now run the quality step of CheckM checkm qa
and make it print the output in a TSV table instead of the console. In this step, we can ask CheckM to give us more parameters, like contig number and length.
Ideally, we would like to get only one contig per bin, with a length similar to the genome size of the corresponding taxa. Since this scenario is complicated to obtain, we can use parameters showing how good our assembly is. Here are some of the most common metrics: If we arrange our contigs by size, from larger to smaller, and divide the whole sequence in half, N50 is the size of the smallest contig in the half that has the larger contigs; and L50 is the number of contigs in this half of the sequence. So we want big N50 and small L50 values for our genomes. Read What is N50?.
To get the table with these extra parameters, we need to specify the file of the markers that CheckM used in the previous step, Bacteria.ms, the name of the output file we want, quality_JP4D.tsv, that we want a table --tab_table, and the option number 2 -o 2 is to ask for the extra parameters printed on the table.
$ checkm qa CHECKM/Bacteria.ms CHECKM/ --file CHECKM/quality_JP4D.tsv --tab_table -o 2
The table we just made looks like this. This will be very useful when you need to document or communicate your work.
The question of how much contamination we can tolerate and how much completeness we need certainly depends on the scientific question being tackled, but in the CheckM paper, there are some parameters that we can follow.
Exercise 1: Discuss the quality of the obtained MAGs
Fill in the blanks to complete the code you need to download the
quality_JP4D.tsvto your local computer:____ dcuser____ec2-18-207-132-236.compute-1.amazonaws.com____/home/dcuser/dc_workshop/mags/CHECKM/quality_JP4D.tsv ____Solution
In a terminal that is standing on your local computer, do:
$ scp dcuser@ec2-18-207-132-236.compute-1.amazonaws.com:/home/dcuser/dc_workshop/mags/CHECKM/quality_JP4D.tsv <the destination directory of your choice>Then open the table in a spreadsheet and discuss with your team which of the parameters in the table you find useful.
Key Points
Metagenome-Assembled Genomes (MAGs) sometimes are obtained from curated contigs grouped into bins.
Use MAXBIN to assign the contigs to bins of different taxa.
Use CheckM to evaluate the quality of each Metagenomics-Assembled Genome.